May. 3, 2023 - What You Need to Know about the New Parkinson’s Biomarker
Mostrando postagens com marcador alfa-sinucleína. Mostrar todas as postagens
Mostrando postagens com marcador alfa-sinucleína. Mostrar todas as postagens
quinta-feira, 4 de maio de 2023
segunda-feira, 17 de abril de 2023
Parkinson e alfa-sinucleína

170423 - O que é alfa-sinucleína?
A
alfa-sinucleína é uma proteína extremamente abundante em nossos
cérebros; constituindo cerca de 1% de todas as proteínas que
flutuam em cada neurônio (um dos principais tipos de células do
cérebro). As proteínas compõem a maior parte das vias biológicas
que ocorrem dentro de cada neurônio e permitem que nossos cérebros
funcionem. Para que cada proteína funcione corretamente, elas devem
ser fabricadas corretamente.
Em neurônios saudáveis, a
alfa-sinucleína corretamente construída é normalmente encontrada
apenas dentro da superfície da membrana do neurônio, bem como nas
pontas dos ramos que se estendem dos neurônios (em estruturas
chamadas terminais pré-sinápticos, que são essenciais para passar
as mensagens químicas entre os neurônios).
Por que a
alfa-sinucleína é relevante para o Parkinson?
Cinco mutações
genéticas no gene da alfa-sinucleína foram identificadas como um
risco aumentado de Parkinson - elas representam 10-20% dos casos de
Parkinson. Então, do ponto de vista genético, a alfa-sinucleína
está associada ao Parkinson; mas também está associado a nível de
proteína.
No cérebro de muitas pessoas com Parkinson,
descobriu-se que algumas proteínas alfa-sinucleína são dobradas de
maneira desordenada. Essas versões incorretamente construídas de
alfa-sinucleína se agrupam em agregados chamados de “corpos de
Lewy”. Os corpos de Lewy são aglomerados circulares de
alfa-sinucleína (e outras proteínas) encontrados no cérebro de
pessoas com Parkinson. Eles são abundantes em áreas do cérebro que
sofreram perda celular, como a região que contém neurônios
produtores de dopamina.
Alfa-sinucleína e formas de corpos de
Lewy
Não sabemos o que causa a formação dos corpos de Lewy, mas
há muitas evidências que apóiam a ideia de que a alfa-sinucleína
é transmitida entre os neurônios. Uma vez lá dentro, a
alfa-sinucleína ‘semeia’ a formação de novos corpos de Lewy
dentro do novo neurônio, e é assim que se acredita que a doença
progride.
Podemos impedir a aglomeração de alfa-sinucleína
e os corpos de Lewy de se desenvolverem e se espalharem?
Esta é
uma pergunta muito interessante e que está sendo feita e investigada
por pesquisadores de todo o mundo.
Uma área de pesquisa é a
de vacinas que têm como alvo a alfa-sinucleína; a ideia é que
essas vacinas irão capturar e remover a alfa-sinucleína que está
sendo transmitida entre as células e, assim, interromper ou pelo
menos retardar a progressão do Parkinson.
Outras áreas de
pesquisa estão focadas em medicamentos que inibem a formação de
grumos de alfa-sinucleína.
Uma área de pesquisa em que a
Cura de Parkinson está envolvida é a do medicamento, ambroxol, que
demonstrou melhorar a eliminação de resíduos das células,
incluindo a alfa-sinucleína mal dobrada. Original em inglês,
tradução Google, revisão Hugo. Fonte: Cureparkinsons.
O modelo do cérebro primeiro versus o corpo primeiro da doença de Parkinson em comparação com modelos alternativos
2023 Apr 16 - Resumo
A origem final dos distúrbios dos corpos de Lewy, incluindo a doença de Parkinson (DP) e a demência com corpos de Lewy (DLB - Dementia with Lewy bodies), ainda não é completamente compreendida. Embora um grande número de mecanismos patogênicos tenha sido implicado, evidências acumuladas sustentam que a agregação e a propagação de neurônio a neurônio da alfa-sinucleína podem ser a característica central desses distúrbios. O modelo de doença sinucleína, origem e conectoma (SOC - synuclein, origin, and connectome) dos distúrbios do corpo de Lewy foi introduzido recentemente. Este modelo baseia-se na hipótese de que, na maioria dos pacientes, a primeira patologia da alfa-sinucleína surge em um único local e se espalha a partir daí. Os locais de origem mais comuns são o sistema nervoso entérico e o sistema olfatório. O modelo SOC prevê que a patologia gut-first (intestino primeiro) leva a um subtipo clínico de body-first (corpo primeiro) caracterizado por sintomas autonômicos prodrômicos e distúrbio comportamental do sono REM. Em contraste, a patologia olfativa leva a um subtipo cerebral com menos sintomas não motores antes do diagnóstico. O modelo SOC prevê ainda que os pacientes que priorizam o corpo são mais velhos, desenvolvem mais comumente degeneração dopaminérgica simétrica e apresentam maior risco de demência em comparação com pacientes que priorizam o cérebro. Nesta revisão, o modelo SOC é explicado e comparado a modelos alternativos da patogênese dos distúrbios do corpo de Lewy, incluindo o sistema de estadiamento de Braak e o Sistema de Estadiamento Unificado para distúrbios do corpo de Lewy. Evidência post-mortem de bancos de cérebro e dados de imagem clínica de perda dopaminérgica e simpática cardíaca é revisada. Conclui-se que esses conjuntos de dados parecem ser mais compatíveis com o modelo SOC do que com os modelos alternativos de doenças de corpos de Lewy. Original em inglês, tradução Google, revisão Hugo. Fonte: Pub Med.
sábado, 1 de abril de 2023
As lipoproteínas do líquido cefalorraquidiano inibem a agregação de α-sinucleína ao interagir com espécies oligoméricas em ensaios de amplificação de sementes
terça-feira, 28 de março de 2023
Fosforilação da sinucleína: patogênica ou fisiológica?
28 March 2023 - Apesar de vários caminhos terapêuticos para o manejo dos sintomas na doença de Parkinson (DP), ainda não há tratamento para o processo degenerativo subjacente. Como resultado, a doença continua a progredir, tornando-se refratária ao tratamento e resultando em incapacidade grave. Para resolver isso, os pesquisadores se concentraram nas proteínas implicadas na DP pela genética humana e, em particular, na proteína α-sinucleína, que se acumula no sistema nervoso de pacientes com DP e nos distúrbios relacionados demência com corpos de Lewy (LBD) e sistemas múltiplos atrofia (MSA). O desdobramento incorreto da sinucleína e a herança dominante da DP devido a mutações na sinucleína e na quinase de repetição rica em leucina 2 (LRRK2) levaram à hipótese de que a doença envolve um ganho de função anormal. Como corolário, a função normal dessas proteínas foi considerada irrelevante para a doença. É verdade que a perda de sinucleína não causa a DP: camundongos knockout sem todas as três isoformas de sinucleína não apresentam perda celular ou sintomas motores parkinsonianos1,2. No entanto, a degeneração surge no contexto da função normal, tornando-se essencial entender o papel fisiológico adaptativo dessas proteínas, que permanece indefinido.
Dettmer, Selkoe e colegas agora abordam o papel
fisiológico de uma modificação pós-traducional associada à
sinucleína + patologia Lewy característica de PD3. Considerada
específica para a DP, a fosforilação da α-sinucleína em Ser-129
(pSer-129)4 tem sido amplamente utilizada para avaliar a magnitude e
a extensão da degeneração em modelos animais, bem como na condição
humana. Em geral, tem sido considerado um marcador da patologia de
Lewy, e o papel de pSer-129 na patogênese tem sido objeto de debate.
Vários relatórios implicaram a modificação na toxicidade5,6,7,
mas outro sugeriu um efeito inibitório na fibrilação da
sinucleína8. E outros estudos não encontraram nenhum efeito da
modificação9,10,11. No entanto, nenhum desses estudos se concentrou
em um papel fisiológico potencial para pSer-129, embora trabalhos
anteriores tenham detectado essa modificação no cérebro normal com
alguma regulação por um forte estímulo sensorial12,13. Além
disso, esse resíduo é altamente conservado na α-sinucleína, mas
não nas isoformas β ou γ, sugerindo um papel adaptativo
específico.
No presente estudo, Ramalingam et al. mostram que
a atividade neural aumenta pSer-129 ~3 vezes na cultura primária,
sem alteração na α-sinucleína total3. Essa indução requer
potenciais de ação e transmissão sináptica, indicando que a
atividade da rede é responsável. Os autores também observaram um
aumento mais modesto em extratos de cérebro após enriquecimento
ambiental, apoiando a relevância da atividade na indução de
pSer-129 in vivo. Além disso, a atividade-dependência da
fosforilação parece específica para Ser129, não para outros
sítios conhecidos de fosforilação na α-sinucleína. Para entender
como a atividade aumenta pSer-129, os autores identificam a quinase 2
tipo polo (Plk2) como responsável. O Ca++ geralmente medeia o efeito
da atividade e os autores descobriram que os canais de Ca++
controlados por voltagem são importantes para a fosforilação, mas
Plk2 não é conhecido por responder ao Ca++. Os autores buscaram,
assim, um potencial ativador a montante, identificando a fosfatase
calcineurina sensível ao Ca++. A inibição da calcineurina reduz a
pSer-129 na mesma proporção que a inibição da Plk2, sem efeitos
aditivos, sugerindo que atuam na mesma via. É importante ressaltar
que a calcineurina tem um papel importante na liberação de
neurotransmissores e a α-sinucleína é altamente pré-sináptica.
Para determinar onde ocorre a fosforilação da Ser-129 na célula, os autores fracionam os neurônios após a indução da atividade neural e descobrem que a maioria (mas não todas) da α-sinucleína modificada se associa às membranas. A imunocoloração confirma a expressão de pSer-129 nos botões pré-sinápticos. No entanto, talvez o resultado mais notável seja o aumento da fosforilação in vitro com Plk2 na presença de lipossomas. A α-sinucleína é uma proteína de membrana periférica que provavelmente se associa a vesículas sinápticas e a associação de membrana com membranas artificiais in vitro requer o N-terminal altamente conservado, com sete repetições de 11 aminoácidos que formam uma α-hélice na ligação à membrana14. pSer-129 ocorre no terminal C menos altamente conservado, e o aumento na fosforilação de Ser-129 na associação de membrana pode ocorrer por deslocamento do terminal C de uma interação intramolecular com as repetições de ligação à membrana N-terminal. A associação de membrana pode influenciar de forma semelhante a ligação do terminal C da α-sinucleína ao v-SNARE VAMP21. Uma vez que a α-sinucleína se liga com baixa afinidade às membranas pré-sinápticas e liga e desliga dinamicamente com exocitose e reciclagem15, a associação de membrana, bem como a ativação de Plk pela calcineurina, pode regular a fosforilação de Ser-129. No entanto, a fosforilação em Ser-129 não parece influenciar a interação da α-sinucleína com as membranas, de modo que a associação de membrana parece promover a fosforilação em Ser-129, e não vice-versa. Isso presumivelmente explica o aumento da localização pré-sináptica da sinucleína fosforilada em Ser-129 que os autores também demonstraram.
Finalmente, Ramalingam et al. (2023) abordam o
papel funcional da fosforilação de Ser-1293, uma tarefa difícil
devido ao papel pouco claro da α-sinucleína na neurotransmissão
estudada em camundongos KO1,2. Apresentando WT e α-sinucleína
mutante que não pode sofrer fosforilação em Ser-129 em neurônios
KO de α-sinucleína, eles descobrem que, em relação a WT, o
mutante reduz a amplitude das correntes pós-sinápticas excitatórias
(EPSCs) e aumenta a amplitude das correntes pós-sinápticas
inibitórias ( IPSCs), sem alteração em sua frequência. Isso é
surpreendente porque, como uma proteína pré-sináptica, seria
esperado que a α-sinucleína influenciasse a frequência de eventos.
Uma mudança na amplitude é geralmente considerada como refletindo
uma mudança nos receptores pós-sinápticos, onde há relativamente
pouca sinucleína. Alternativamente, uma mudança na amplitude pode
refletir o preenchimento alterado da vesícula sináptica com
neurotransmissor e a sinucleína pode afetar o vazamento das
vesículas sinápticas, mas a ausência completa de sinucleína não
demonstrou afetar nenhuma dessas propriedades. Os autores também
examinaram a liberação evocada em fatias do hipocampo e encontraram
um efeito modesto na razão de pulsos pareados e um efeito maior na
depressão sináptica, parâmetros mais claramente associados à
função pré-sináptica, mas novamente sem grandes alterações em
camundongos KO sinucleína16.
Em resumo, os autores fornecem
evidências claras para a regulação fisiológica da fosforilação
da α-sinucleína em Ser-129 pela atividade neural, indicando um
papel além da patologia de Lewy. Além de Ca++ agir através da
calcineurina e Plk2, a associação de membrana parece regular
pSer-129, mas pSer-129 aparentemente não influencia a associação
de membrana. A modificação também parece afetar a
neurotransmissão, embora o mecanismo permaneça obscuro. Talvez o
mais interessante, os resultados sugerem um importante papel
regulador para o terminal C da α-sinucleína menos conservado. As
mutações pontuais que causam a DP ocorrem dentro de uma pequena
região nas repetições de ligação à membrana N-terminal, mas a
regulação no C-terminal pode controlar a função da sinucleína na
degeneração, bem como a fisiologia. Original em inglês, tradução
Google, revisão Hugo (assunto muito difícil sobre a
alfa-sinucleína). Fonte: Nature.
segunda-feira, 13 de março de 2023
segunda-feira, 26 de dezembro de 2022
A trilha oculta célula a célula dos agregados de α-sinucleína
2022 22 de dezembro - resumo
O acúmulo progressivo de agregados insolúveis da proteína pré-sináptica alfa-sinucleína (α-Syn) é uma marca registrada de distúrbios neurodegenerativos, incluindo doença de Parkinson (DP), Atrofia de Múltiplos Sistemas e Demência com Corpos de Lewy, comumente referidos como sinucleinopatias. Apesar do progresso considerável na biologia estrutural desses agregados, os mecanismos moleculares que medeiam sua transmissão, propagação e neurotoxicidade célula a célula permanecem apenas parcialmente compreendidos. Numerosos estudos destacaram a propagação espaço-temporal estereotipada de agregados α-Syn patológicos em diferentes tecidos e regiões cerebrais conectadas anatomicamente ao longo do tempo. Evidências experimentais de vários modelos celulares e animais indicam que a transferência de α-Syn ocorre em duas etapas definidas: a liberação de espécies patogênicas de α-Syn de células infectadas e sua captação por vias endocíticas passivas ou ativas. Uma vez que os agregados α-Syn tenham sido internalizados, pouco se sabe sobre o que impulsiona sua toxicidade ou como eles interagem com a proteína endógena para promover seu dobramento incorreto e subsequente agregação. Da mesma forma, fatores genéticos desconhecidos modulam diferentes respostas celulares à agregação e acúmulo de espécies α-Syn patogênicas. Aqui discutimos a compreensão atual dos fenômenos moleculares associados à propagação intercelular de sementes α-Syn patogênicas e resumimos as evidências que sustentam a hipótese de transmissão. Compreender os mecanismos moleculares envolvidos na transmissão de agregados α-Syn é essencial para desenvolver novas terapêuticas direcionadas contra a DP e sinucleinopatias relacionadas. Original em inglês, tradução Google, revisão Hugo. Fonte: Pubmed.
sábado, 17 de dezembro de 2022
À medida que o Parkinson progride, as fibrilas de sinucleína mudam de forma?
16 Dec 2022 - Fibrilas de α-sinucleína podem ser o denominador comum entre as sinucleinopatias, mas nem todas mapeiam para o mesmo projeto. As fibrilas de pessoas com atrofia de múltiplos sistemas dobram-se de maneira diferente daquelas encontradas no Parkinson, na demência da doença de Parkinson e na demência com corpos de Lewy. Se fibrilas únicas podem se formar em diferentes doenças, conformações específicas também podem surgir em diferentes estágios de uma única doença? Sim, dizem os cientistas liderados por Dan Li na Shanghai Jiao Tong University e Jian Wang na Fudan University, também em Shanghai.
Fibras de sinucleína
amplificadas a partir do líquido cefalorraquidiano.
Uma
conformação é abundante em todos os estágios do Parkinson.
Um
confôrmero menor apenas do LCR (líquido cefalorraquidiano) em
estágio avançado semeia novas fibrilas de forma potente.
Na Structure de 12
de dezembro, eles relatam um conformador de uma pessoa com Parkinson
em estágio avançado que não foi detectado em outras pessoas em
estágio intermediário ou pré-clínico da doença. As descobertas
sugerem que as fibrilas de α-sinucleína sofrem uma transição
conformacional à medida que a doença progride. Os cientistas se
perguntam se outros amilóides, incluindo Aβ e fibrilas tau, também
podem mudar de forma ao longo do tempo.
Deslocamento de
Sinucleínas. No Parkinson, os polimorfos de α-sinucleína mais
abundantes são os mesmos ao longo da progressão da doença.
Polimorfos menores, no entanto, são diferentes. A variante mais
tóxica para os neurônios surge no estágio avançado da DP.
[Cortesia de Fan et al., Structure, 2022.]
Para ser claro, os
primeiros autores conjuntos Yun Fan, da Universidade de Fudan, e
Yunpeng Sun, do Instituto de Química Orgânica de Xangai, não
isolaram fibrilas do tecido cerebral, como estudos recentes de
crio-EM foram capazes de fazer. Eles usaram uma técnica desenvolvida
pelo grupo de Claudio Soto na Universidade do Texas para estimular o
crescimento de protofibrilas usando o líquido cefalorraquidiano como
semente (Shahnawaz et al., 2020).
Eles testaram o LCR de
quatro controles saudáveis, de uma pessoa com DP pré-clínica,
quatro com DP em estágio intermediário e uma pessoa com doença em
estágio avançado. O método de amplificação cíclica de
dobramento incorreto de proteínas (PMCA - protein misfolding cyclic amplification - amplificação cíclica de dobramento incorreto de proteínas) não produziu fibrilas do
CSF de controle, mas todas as seis amostras de DP semearam agregados
de sinucleína. Quando os cientistas examinaram essas fibrilas por
crio-EM, eles encontraram um grande polimorfo em todos os estágios
da doença (veja a imagem abaixo). Dobra-se de forma semelhante às
fibrilas de sinucleína feitas in vitro (Guerrero-Ferreira et al.,
2019).

Transformando Polimorfos. Os principais polimorfos de sinucleína de todos os estágios da doença eram idênticos (esquerda), compreendendo duas hélices canhotas (roxo e dourado) que se enrolam uma na outra. O polimorfo menor da DP de estágio intermediário (centro) tinha protofibrilas quase idênticas (marrom e azul) às das formas principais, mas elas se envolviam em uma conformação diferente. O polimorfo menor da DP em estágio avançado (à direita) tinha uma dobra única; duas protofibrilas idênticas (ciano) se abraçaram para formar fibrilas. [Cortesia de Fan et al., Structure, 2022]
Os polimorfos menores eram mais
interessantes. No estágio intermediário da DP, eles eram muito
semelhantes aos principais polimorfos, adotando as mesmas dobras, mas
envolvendo-se de maneira diferente para formar fibrilas (veja a
imagem acima).
A forma menor da DP tardia era completamente
diferente. Duplas de uma única protofibrila abraçadas para formar
fibrilas, formando uma estrutura que não havia sido relatada antes.
Ainda assim, essas protofibrilas adotaram dobras substancialmente
semelhantes às observadas nas fibrilas de Li feitas anteriormente in
vitro e com dobras encontradas na sinucleína isolada do cérebro de
pessoas com atrofia de múltiplos sistemas (Li et al., 2018; notícias
de março de 2020). As diferenças incluem um terminal C mais
alongado na variante amplificada em estágio avançado versus a
estrutura MSA e um gancho β nos resíduos 59-72 na protofibrila PMCA
com cadeias laterais invertidas em 180 graus (veja a imagem abaixo).
Os autores observam que as fibrilas MSA incorporam modificações
pós-traducionais e cofatores misteriosos que podem não ser
totalmente capturados pela amplificação de PMCA, portanto, podem
não representar totalmente o conformador pai que se esconde no
cérebro.

PD á la MSA? O polimorfo menor amplificado do LCR de DP em estágio avançado (ciano) dobra-se de forma semelhante (canto superior esquerdo) para protofibrilas de sinucleína isoladas de tecido cerebral MSA (cinza) ou amplificadas de fibrilas MSA (roxo). Visualizações ampliadas i, ii e iii mostram as principais diferenças. [Cortesia de Fan et al., Structure, 2022.]
O polimorfo da DP em estágio avançado revela algo sobre a progressão da doença? Curiosamente, a amostra de PMCA em estágio avançado foi a mais tóxica para os neurônios em cultura, induzindo fortemente a fosforilação da α-sinucleína endógena e semeando novas fibrilas. Uma vez que esta amostra continha 27 por cento de polimorfo menor, contra 8 por cento a 17 por cento para as amostras pré e intermediárias, os autores atribuíram esta toxicidade ao polimorfo menor de estágio avançado.—Tom Fagan. Original em inglês, tradução Google, revisão Hugo. Fonte: Alzforum.
sábado, 5 de novembro de 2022
domingo, 9 de outubro de 2022
Combinação de Biomarcadores Associados a Previsões Mais Fortes de Progressão na Doença de Parkinson
October 8, 2022 - Os resultados de um estudo sobre a doença de Parkinson revelaram que a combinação de biomarcadores sanguíneos, além de medidas clínicas com modelagem de prognósticos, está associada a uma previsão mais vital na progressão da doença.
Nirosen Vijiaratnam, MBBS,
pesquisador acadêmico, Departamento de Neurociências Clínicas e do
Movimento, University College London, UCL Queen Square Institute of
Neurology
Nirosen Vijiaratnam, MBBS
Um estudo recente em
291 pacientes com doença de Parkinson (DP) mostrou que uma
combinação de luz de neurofilamento sérico (NfL - serum
neurofilament light), resultados clínicos e status genético dos
pacientes pode ajudar na previsão da progressão da DP.1 Ter
previsões mais precisas de progressão em pacientes com DP pode
melhorar a seleção para ensaios clínicos.
Não só o NfL
basal foi associado ao estado cognitivo basal, mas também predisse
um tempo mais curto para demência (HR, 2,64), instabilidade postural
(HR, 1,32) e morte (HR, 1,89).1 Em comparação, apolipoproteína E
(APOE - apolipoprotein E) ε4 o status foi associado à progressão
para demência (HR 3,12; IC 95%, 1,63-6,00). As variáveis
genéticas e os níveis de NfL previram uma progressão
desfavorável em contraste semelhante aos preditores clínicos.
Ao
combinar dados de NfL, clínicos e genéticos, os dados produziram
uma previsão mais forte de resultados desfavoráveis, indicados por
uma área sob a curva (AUC = area under the curve) de 0,84, em
comparação com idade e sexo, que tiveram uma AUC de 0,74 (P = .
0103). O NfL sérico em combinação com variáveis genéticas
como glicocerebrosidase (GBA) e APOE forneceu uma melhor previsão de
vários aspectos da progressão da DP na modelagem prognóstica do
que apenas com medidas clínicas.
O investigador principal
Nirosen Vijiaratnam, MBBS, pesquisador acadêmico, Departamento de
Neurociências Clínicas e do Movimento, University College London,
UCL Queen Square Institute of Neurology e colegas observaram que eles
“classificaram os pacientes como tendo um resultado favorável ou
desfavorável com base em um resultado previamente validado modelo e
explorou como os biomarcadores sanguíneos se comparam com as
variáveis clínicas na distinção de fenótipos
prognósticos”.
As alterações do sono experimentadas pelos
pacientes em infusão de apomorfina foram indicadas por pontuações
no Índice de Gravidade da Insônia e na Escala de Melhoria de
Impressão Clínica Global.
Os pacientes com DP incluídos na
análise foram recrutados a partir do estudo de observação Tracking
Parkinson's. Os pacientes inscritos no estudo preencheram os
critérios do Queen Square Brain Bank e tiveram neuroimagem de
suporte. Os pacientes também tinham que estar dentro de 3,5 anos
após o diagnóstico no recrutamento, o que incluiu pacientes virgens
de tratamento e tratados com idades entre 18 e 90 anos.
Foi
necessário um seguimento mínimo de 2,5 anos para que os pacientes
fossem selecionados para a análise de NfL. Além disso, para
facilitar uma análise sobre se a NfL pode ajudar a discriminar a DP
típica com alto índice de certeza diagnóstica (maior que 95%), de
uma amostra equivalente de casos com características clínicas
atípicas com menor índice de certeza diagnóstica (menos de 80 %),
outros critérios de seleção foram aplicados em sua avaliação
clínica de 2,5 anos. As limitações do estudo foram a falta de
avaliação no estado de medicação OFF e a falta de confirmação
diagnóstica neuropatológica na coorte.
Outros achados foram
que os escores basais de MoCA e SF foram associados aos níveis de
NfL, que foram consistentes com outros estudos que exploraram a
função cognitiva global,2 e destacaram o valor de testes
neuropsicológicos mais detalhados. Além disso, não houve diferença
significativa nos níveis de NfL ao comparar pacientes com status
APOE ε4 heterozigoto ou homozigoto para aqueles que não o fizeram.
Embora a capacidade preditiva do status APOE ε4 no desenvolvimento
de demência e progressão cognitiva tenha sido confirmada,
observaram Vijiaratnam et al.1
Houve um efeito principal
negativo de níveis basais mais altos de NfL nos escores de
progressão na modelagem mista, que era consistente com os níveis de
NfL atingindo o pico antes do início de características clínicas
apreciáveis. conforme determinado por mudanças no MoCA, os
pesquisadores identificaram uma capacidade preditiva significativa
para o desenvolvimento precoce de demência. Isso apoiou um estudo
anterior que sugeriu que a revelação de NfL é melhor na previsão
do desenvolvimento de demência do que o comprometimento cognitivo
leve.4
Com base nos resultados, a capacidade da NFL de prever a progressão cognitiva pode ser explicada através da previsão de progressão mais precisa, refletindo a magnitude da deposição de alfa-sinucleína e disfunção anatômica presente. Status da NfL no momento do recrutamento”, escreveram Vijiaratnam e colegas. Original em inglês, tradução Google, revisão Hugo. Fonte: Neurology live.
sábado, 8 de outubro de 2022
'É extremamente complexo': os cientistas que trabalham para derrotar o Parkinson
Pesquisa em células produtoras de dopamina e proteínas nocivas entre os esforços para encontrar um tratamento de longo alcance
Fri 7 Oct 2022 - Foi enquanto assistia ao Desafio Universitário que o médico suspeitou pela primeira vez de algo errado com Jeremy Paxman. Normalmente altamente animado, o apresentador de TV estava menos efusivo e exuberante do que o habitual. Ele havia adquirido o que os especialistas da área chamam de “máscara de Parkinson”.
Paxman foi formalmente
diagnosticado com doença de Parkinson no hospital depois que ele
desmaiou enquanto passeava com seu cachorro e se viu no hospital. Lá,
lembrou Paxman em um documentário da ITV, o médico entrou e disse:
“Acho que você tem Parkinson”. Para Paxman, pelo menos, a
notícia veio do nada.
O Parkinson foi descrito pela primeira
vez em textos médicos há mais de 200 anos, mas ainda não há cura.
É uma condição comum, principalmente em pessoas com mais de 50
anos. Cerca de 1 em cada 37 pessoas no Reino Unido será
diagnosticada em algum momento de sua vida. Os medicamentos
existentes visam controlar os sintomas dos pacientes, em vez de
retardar ou interromper a progressão da doença. Mas os cientistas
fizeram progressos na compreensão do distúrbio neurodegenerativo. A
esperança agora é que as terapias revolucionárias estejam
finalmente no horizonte.
“Parkinson é uma condição
extremamente complexa e provavelmente não há cura única”, diz
Katherine Fletcher, gerente de comunicações de pesquisa do
Parkinson’s UK. “É a perda progressiva de células produtoras de
dopamina no cérebro. Se você quiser retardar ou parar a condição,
de alguma forma você precisa proteger essas células ou talvez até
regenerar essas células no cérebro. Esse é o objetivo final.”
Por
que as células cerebrais morrem no Parkinson ainda é desconhecida.
A condição atinge uma região do cérebro chamada substância
negra, onde os neurônios produzem uma substância química chamada
dopamina. A perda dessas células cerebrais faz com que a dopamina
caia, e isso impulsiona a maioria dos problemas que o paciente
experimenta. Não é um declínio rápido: normalmente, os pacientes
só percebem os sintomas quando cerca de 80% das células nervosas da
substância negra falharam.
Embora existam sintomas comuns, o
Parkinson afeta as pessoas de maneira muito diferente. Os problemas
mais proeminentes são tremores, dificuldade em andar e músculos
rígidos, mas mais de 40 sintomas são reconhecidos. Perda de sono,
problemas de equilíbrio, memória fraca, ansiedade, depressão,
perda de olfato, constipação – a lista continua. Os músculos
faciais são frequentemente afetados, levando à “máscara de
Parkinson”. O impacto não deve ser subestimado: as pessoas podem
parecer vazias e sem emoção, independentemente do que estão
sentindo por dentro.
O principal tratamento
para o Parkinson visa aumentar os níveis de dopamina no cérebro. A
droga, levodopa, tende a ser tomada com outros medicamentos para
fazê-la funcionar por mais tempo com menos efeitos colaterais. No
entanto, nem todos os pacientes respondem aos medicamentos e, em
alguns casos, os médicos realizam cirurgias para inserir eletrodos
profundamente no cérebro. Pulsos elétricos disparados de um
implante no peito podem aliviar tremores e outros sintomas.
Terapias
muito mais radicais estão em ensaios clínicos. Médicos japoneses
estão monitorando sete pacientes que tiveram milhões de neurônios
produtores de dopamina implantados nas regiões mais afetadas do
cérebro. Os neurônios foram feitos reprogramando células-tronco em
laboratório. Os resultados do julgamento devem sair em
breve.
Outras abordagens visam proteínas problemáticas
envolvidas no distúrbio. Um, chamado alfa-sinucleína, é encontrado
em aglomerados dentro dos neurônios de pessoas com Parkinson. Muitos
pesquisadores acreditam que isso contribui para a doença e
impulsiona sua disseminação pelo cérebro. Mas enquanto os
cientistas tentaram limpar a alfa-sinucleína com infusões de
anticorpos sintéticos, os testes até agora não mostraram nenhum
benefício.
O trabalho deste ano de Alice Chen-Plotkin,
professora de neurologia da Universidade da Pensilvânia, aponta para
outra rota potencial para o tratamento do Parkinson. Sua equipe
descobriu que a alfa-sinucleína funciona com uma proteína chamada
GPNMB para entrar nos neurônios. Eles agora estão analisando se a
redução dos níveis de GPNMB afeta a disseminação da
alfa-sinucleína. “Se este for o caso, esperamos que a diminuição
dos níveis de GPNMB, ou o bloqueio de sua capacidade de interagir
com a alfa-sinucleína, possa impedir que a alfa-sinucleína anormal
se espalhe de áreas afetadas do cérebro para áreas saudáveis do
cérebro em humanos." ela disse.
Outros esforços estão
focados no papel das mitocôndrias, as pequenas estruturas
semelhantes a baterias que ficam dentro das células vivas. Estudos
sugerem que o mau funcionamento das mitocôndrias é um importante
fator de Parkinson em estágio inicial. “Há evidências muito
fortes de disfunção mitocondrial em todos os diferentes tipos de
Parkinson”, disse Oliver Bandmann, professor de neurologia de
distúrbios do movimento da Universidade de Sheffield.
Um
estudo recente liderado por Bandmann analisou se um medicamento que
aumenta as mitocôndrias usado para tratar uma doença hepática rara
poderia ser reaproveitado para ajudar pessoas com Parkinson. O estudo
descobriu que a droga, UDCA, era segura e melhorava a função das
mitocôndrias no cérebro das pessoas. A análise sofisticada da
marcha dos participantes do estudo também encontrou sinais
promissores de melhora, embora Bandmann diga que um estudo maior e
mais longo é necessário para confirmar qualquer benefício. “Na
minha opinião, é muito provável que pacientes com distúrbios
neurodegenerativos acabem em um coquetel de drogas – drogas que
resgatam a função mitocondrial, que reduzem a agregação de
alfa-sinucleína e assim por diante”, disse ele.
Enquanto os
esforços para descobrir novos medicamentos continuam, os pacientes
estão sendo incentivados a se exercitar se puderem, em meio a
evidências de que a atividade física pode ajudar as pessoas a
controlar seus sintomas e até proteger o cérebro. “Parece
realmente benéfico, por isso incentivamos as pessoas a se
exercitarem”, disse Fletcher. Original em inglês, tradução
Google, revisão Hugo. Fonte: The Guardian.
quinta-feira, 22 de setembro de 2022
Parkinson: sabemos mais sobre a propagação da doença no cérebro
220922 - Décadas de pesquisa sobre a doença de Parkinson implicaram agregados de proteínas neuronais na disseminação da doença. Agora, os pesquisadores identificaram a exocitose lisossomal como responsável pela liberação desses agregados e sua reprodução patogênica no cérebro.
Embora existam tratamentos
para aliviar certas anormalidades do movimento características da
doença de Parkinson, nenhum deles atualmente pode interromper a
progressão desse distúrbio neurológico. O motivo é a falta de
conhecimento sobre esse processo.
Atualização sobre
resultados anteriores
As últimas décadas de pesquisa sobre a
doença de Parkinson mostraram que a morte de neurônios em pacientes
está associada à liberação de agregados patogênicos da proteína
neuronal alfa-sinucleína (αSyn) no espaço extracelular. Os
pesquisadores levantaram a hipótese de que essa liberação de
agregados causou a disseminação em cadeia da doença de um neurônio
para outro.
Experimentos anteriores em camundongos e primatas
não humanos mostraram que a injeção desses agregados no cérebro
pode desencadear essa disseminação, bem como a neurodegeneração
do tipo Parkinson. No entanto, como os neurônios transmitem esses
agregados para outros neurônios nunca foi detalhado antes.
A
exocitose lisossomal libera espécies patogênicas de alfa-sinucleína
(αSyn) dos neurônios para o espaço extracelular. Essa liberação
é regulada pela atividade neuronal e pelo cálcio citosólico. ©
Xie, Y.X., Naseri, N.N., Fels, J. et alii. Nat comum.
O
processo de exocitose lisossomal explicado
Os pesquisadores da
Weill Cornell Medicine usaram modelos de camundongos da doença de
Parkinson, onde os agregados de αSyn são produzidos dentro dos
neurônios. Seus resultados, publicados na Nature Communications,
mostram que esses agregados se acumulam nas vesículas celulares: os
lisossomos. Estes normalmente contêm enzimas capazes de digerir e
reciclar moléculas, como resíduos celulares.
Mas o processo
não funciona bem com agregados de αSyn, que muitas vezes estão
ligados em uma estrutura em camadas muito apertada chamada
"amilóide". Por um processo de exocitose lisossomal, o
lisossomo se move em direção à membrana celular e se funde com
ela, e seu conteúdo é então descarregado para fora da célula.
De
fato, os neurônios liberam agregados de αSyn, que não possuem
membrana e são capazes de se reproduzir. "Esses resultados
propõem a exocitose lisossomal como mecanismo central para a
liberação de proteínas agregadas e resistentes à degradação
pelos neurônios", resumem os autores.
Uma abordagem terapêutica contra doenças neurodegenerativas
A fim de abordar uma cura para a doença de Parkinson, os pesquisadores mostraram em outros experimentos que, ao reduzir a taxa de exocitose lisossomal, eles poderiam reduzir a concentração de agregados propagativos. Manter esses agregados no interior dos lisossomos pode constituir uma estratégia interessante.
Já conhecido, o processo de exocitose lisossomal seria um mecanismo geral para a propagação de agregados proteicos em doenças neurodegenerativas como a doença de Parkinson. Seria, portanto, um alvo geral de escolha para o desenvolvimento de tratamentos e medidas preventivas. Nesse sentido, a equipe de pesquisa está atualmente realizando pesquisas sobre o papel dos lisossomos na doença de Alzheimer. Original em francês, tradução Google, revisão Hugo. Fonte: Futura-sciences.
quarta-feira, 21 de setembro de 2022
Pequenos agregados solúveis de α-sinucleína são as espécies tóxicas na doença de Parkinson
20 September 2022 - Resumo
Agregados solúveis de α-sinucleína variando em tamanho, estrutura e morfologia têm sido intimamente ligados à morte neuronal na doença de Parkinson. No entanto, a heterogeneidade de diferentes espécies de agregados coexistentes torna difícil isolar e estudar suas propriedades tóxicas individuais. Aqui, mostramos um método não perturbativo confiável para separar uma mistura heterogênea de agregados de proteínas por tamanho. Descobrimos que agregados de α-sinucleína de tipo selvagem menores que 200 nm de comprimento, formados durante uma reação de agregação in vitro, causam inflamação e permeabilização de membranas de lipossomas únicos e que agregados maiores são menos tóxicos. O estudo de agregados solúveis extraídos de cérebros humanos post-mortem também revela que esses agregados são semelhantes em tamanho e estrutura aos agregados menores formados em reações de agregação no tubo de ensaio. Além disso, descobrimos que os agregados solúveis presentes nos cérebros da doença de Parkinson são menores, em grande parte menores que 100 nm, e mais inflamatórios em comparação com os agregados maiores presentes nos cérebros de controle. Este estudo sugere que os pequenos agregados não fibrilares de α-sinucleína são as espécies críticas que conduzem a neuroinflamação e a progressão da doença. (segue...) Original em inglês, tradução Google, revisão Hugo. Fonte: Nature.
sexta-feira, 2 de setembro de 2022
LRP1 é um receptor neuronal para captação e disseminação de α-sinucleína
02 September 2022 - Resumo
Fundo
A agregação e disseminação da
proteína α-sinucleína (α-Syn) e a toxicidade neuronal relacionada
são as principais características patológicas da doença de
Parkinson (DP) e da demência por corpos de Lewy (LBD). Estudos
mostraram que espécies patológicas de α-Syn e tau podem se
espalhar de maneira semelhante a príons entre os neurônios, embora
essas duas proteínas tenham papéis patológicos distintos e
contribuam para diferentes doenças neurodegenerativas. É relatado
que a proteína 1 relacionada ao receptor de lipoproteína de baixa
densidade (LRP1) regula a disseminação de proteínas tau; no
entanto, os mecanismos reguladores moleculares de captação e
disseminação de α-Syn, e se também é regulado por LRP1,
permanecem pouco compreendidos. (...)
Resultados
Descobrimos
que a absorção de α-Syn monomérica e oligomérica foi
significativamente reduzida em iPSNs com LRP1-KO em comparação com
os controles WT. A absorção de PFFs de α-Syn também foi inibida
em iPSNs LRP1-KO, embora em uma extensão muito menor em comparação
com monômeros e oligômeros de α-Syn. O bloqueio de resíduos de
lisina em α-Syn diminuiu efetivamente a captação de α-Syn em
iPSNs e o terminal N de α-Syn foi crítico para a captação de
α-Syn mediada por LRP1. Finalmente, nos camundongos Lrp1-nKO, a
disseminação de α-Syn foi significativamente reduzida em
comparação com os irmãos da mesma ninhada
WT.
Conclusões
Identificamos LRP1 como um regulador chave
da captação neuronal de α-Syn, bem como um importante mediador da
disseminação de α-Syn no cérebro. Este estudo fornece novos
conhecimentos sobre o papel fisiológico e patológico de LRP1 no
tráfico de α-Syn e patologia, oferecendo insights para o tratamento
de sinucleinopatias. (segue...) Original em inglês, tradução Google, revisão
Hugo. Fonte: Springer.
quarta-feira, 31 de agosto de 2022
Pesquisadores descobrem onde e por que as proteínas funcionam mal na doença de Parkinson
August 30, 2022 - Resumo:
Os cientistas descobriram
como um acúmulo de proteína prejudicial começa a acontecer dentro
dos neurônios na doença de Parkinson, causando a morte das células
nervosas. Ao analisar como, onde e por que esse acúmulo acontece, o
trabalho fornece uma visão única de um processo biológico
fundamental que leva à doença de Parkinson.
Cientistas
do Francis Crick Institute, UCL e da Universidade de Edimburgo
descobriram como um acúmulo de proteína prejudicial começa a
acontecer dentro dos neurônios na doença de Parkinson, causando a
morte das células nervosas. Ao analisar como, onde e por que esse
acúmulo acontece, o trabalho fornece uma visão única de um
processo biológico fundamental que leva à doença de Parkinson.
O
Parkinson é uma doença neurodegenerativa progressiva que causa
tremores, lentidão dos movimentos, rigidez e pode progredir para
causar problemas cognitivos graves. Afeta cerca de 145.000 pessoas no
Reino Unido, com esse número esperado para aumentar à medida que
mais pessoas vivem mais.
O Parkinson é causado por uma perda
de neurônios em partes específicas do cérebro. Nas células
nervosas afetadas, uma proteína chamada alfa-sinucleína se dobra e
se aglomera em estruturas prejudiciais. Os mecanismos por trás disso
ainda não são totalmente compreendidos.
Em seu artigo,
publicado na Nature Neuroscience hoje (30 de agosto), os
pesquisadores desenvolveram uma nova abordagem sensível para estudar
o que acontece com a alfa-sinucleína durante os estágios iniciais
da doença.
Usando neurônios derivados de células doadas por
pessoas com formas herdadas de Parkinson, bem como de indivíduos
saudáveis, a equipe conseguiu visualizar onde, por que e como essa
proteína começa a se dobrar e se aglomerar dentro das células
nervosas.
A equipe interdisciplinar de neurologistas, químicos
e biólogos estruturais descobriu que a alfa-sinucleína entra em
contato com as membranas, ou revestimentos, de estruturas dentro das
células nervosas. Quando entra em contato com a membrana da
mitocôndria, parte da célula responsável pela geração de
energia, isso desencadeia o dobramento incorreto e a aglomeração da
alfa-sinucleína.
Os aglomerados de proteína então se
acumulam pesadamente na superfície das mitocôndrias danificando sua
superfície, causando buracos na membrana e interferindo na
capacidade das mitocôndrias de criar energia. Eventualmente, isso
leva as mitocôndrias a liberar sinais que causam a morte do
neurônio.
Embora existam vários subtipos de Parkinson, essa
proteína é conhecida por se dobrar e se agrupar em todos os tipos.
Quando os neurônios estão saudáveis, as proteínas mal dobradas
são constantemente limpas e removidas da célula. Pensa-se que, à
medida que as pessoas envelhecem, o processo de remoção desta
proteína prejudicial pode abrandar.
Sonia Gandhi, autora
principal e líder do grupo sênior do Crick, e professora de
neurologia no UCL Queen Square Institute of Neurology, diz: este
processo dentro da célula humana.
"Nosso estudo fornece
informações sobre o que está acontecendo nos estágios iniciais,
quando as proteínas começam a se dobrar, e como elas afetam a saúde
da célula. Isso fornece uma peça importante do quebra-cabeça para
entender os mecanismos biológicos que conduzem ao
Parkinson".
Andrey Abramov, co-autor principal e
professor do UCL Queen Square Institute of Neurology acrescenta:
"Sabemos há algum tempo que as mitocôndrias são anormais na
doença de Parkinson, mas não ficou claro o porquê. induzir danos
mitocondriais e causar a morte celular."
Minee Choi,
primeiro autor e pesquisador sênior de pós-doutorado no Crick, diz:
"Nosso estudo usou neurônios derivados de células retiradas de
pessoas com Parkinson, o que significa que os neurônios com os quais
trabalhamos tinham a mesma composição e características genéticas
das células doentes. em pacientes. Isso significa que podemos estar
mais confiantes de que nosso trabalho reflete o que está acontecendo
nos neurônios do corpo."
Matthew Horrocks, co-autor
principal e professor sênior de Química Biofísica da Universidade
de Edimburgo, acrescenta: danos em amostras biológicas extremamente
complexas. Nossas descobertas lançam luz sobre os primeiros eventos
da doença de Parkinson, processos que só são visíveis usando
abordagens de detecção extremamente sensíveis."
O novo
método inovador desenvolvido pelos pesquisadores também pode ser
usado para estudar como as proteínas se dobram incorretamente em
outras doenças neurodegenerativas e tipos de células, incluindo
células gliais que estão envolvidas em doenças
neurodegenerativas.
A equipe continuará seu trabalho
estudando como o dobramento incorreto de proteínas dentro das
células afeta a função e a saúde da célula. Usando sua nova
abordagem, eles poderão testar novas terapias que visam reduzir o
dobramento incorreto de proteínas e ver se essas terapias podem
devolver a saúde de uma célula doente. Original em inglês,
tradução Google, revisão Hugo. Fonte: Sciencedaily.
terça-feira, 16 de agosto de 2022
domingo, 14 de agosto de 2022
Anticorpos anti-Parkinson de ligação à alfa-sinucleína falham em ensaios clínicos
Sonntag, 14. August 2022 - Alpha-Synuclein-bindende Anti-Parkinson-Antikörper scheitern in klinischen Studien.
Ou seja: vacina não.
sexta-feira, 12 de agosto de 2022
Translocação de espécies distintas de alfa sinucleína do núcleo para processos neuronais durante a diferenciação neuronal
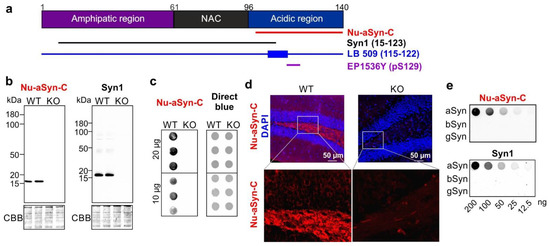
Este é um artigo de acesso aberto distribuído sob a Licença Creative Commons Attribution que permite uso, distribuição e reprodução irrestritos em qualquer meio, desde que o trabalho original seja devidamente citado.
12 August 2022 - Translocation of Distinct Alpha Synuclein Species from the Nucleus to Neuronal Processes during Neuronal Differentiation.
quinta-feira, 4 de agosto de 2022
Um anticorpo monoclonal pode tratar a doença de Parkinson?
August 3, 2022 - Cinpanemab, um anticorpo monoclonal derivado de humanos para α-sinucleína, falha no estudo randomizado de Parkinson.
As
terapias direcionadas à α-sinucleína agregada na doença de
Parkinson (DP) têm sido de grande interesse como um potencial
modificador terapêutico e da doença. O cinpanemab é um anticorpo
monoclonal direcionado ao terminal N derivado de humanos que se liga
à α-sinucleína. Os pesquisadores realizaram um estudo de fase 2,
multicêntrico, duplo-cego, de 52 semanas, focado em 357 pacientes
com DP inicial que foram randomizados 2:1:2:2 para receber placebo ou
doses de cinpanemab de 250 mg, 1.250 mg ou 3.500 mg, entregues
mensalmente. Houve também um período de extensão de tratamento
ativo e cego para a dose por até 112 semanas no total. Em 110 dos
118 participantes testados, foi encontrada α-sinucleína indutora de
agregação no fluido espinhal. O desfecho primário foi a mudança
da linha de base na pontuação total da Escala de Avaliação da
Doença de Parkinson da Movement Disorder Society United (MDS-UPDRS)
nas semanas 52 e 72.
O estudo foi encerrado na semana 72 com
base na falta de eficácia. Em 52 semanas, as alterações no escore
MDS-UPDRS foram de 10,8 pontos no grupo controle, 10,5 pontos com 250
mg de cinpanemab, 11,3 pontos com 1250 mg e 10,9 pontos com 3500 mg.
Os desfechos secundários, incluindo imagens DaT-SPECT na semana 52,
não foram reveladores. Os eventos adversos comuns com cinpanemab
incluíram dor de cabeça, nasofaringite e quedas.
COMENTÁRIO
O
tratamento com anticorpos para a doença de Parkinson, conforme
fornecido neste estudo, pode ser tarde demais para ser eficaz ou não
atingir o alvo certo. A formação de oligômeros de α-sinucleína é
provavelmente um evento precoce e, portanto, pode não ser um alvo
ideal. Os pontos fortes deste estudo foram a inclusão de DP precoce
não tratada e a confirmação da agregação de α-sinucleína em
muitos participantes. Os dados, no entanto, foram claros que
direcionar a α-sinucleína extracelular com um anticorpo direcionado
ao N-terminal não retardou a progressão da doença. O campo
continua precisando de uma nova abordagem terapêutica “fora da
caixa” para abordar a progressão da DP. Original em inglês,
tradução Google, revisão Hugo. Fonte: Jwatch.
Dois ataques e anticorpos monoclonais estão fora de Parkinson precoce?
— A
falha de dois agentes em dois ensaios de fase II pode significar o
fim de uma via de tratamento
August 3, 2022 - Uma renderização
de computador de anticorpos monoclonais.Ensaios de fase II projetados de forma semelhante de dois produtos biológicos direcionados à alfa-sinucleína frustraram as esperanças de drogas de anticorpos monoclonais modificadores da doença no estágio inicial da doença de Parkinson.
No estudo SPARK, o cinpanemab não atingiu nenhum dos endpoints primários para alteração na soma das pontuações nas partes I, II e III da revisão patrocinada pela Movement Disorder Society da pontuação total da Escala Unificada de Avaliação da Doença de Parkinson (MDS-UPDRS), relatou Tien Dam, MD, do desenvolvedor de medicamentos Biogen em Cambridge, Massachusetts, e colegas.
Nenhuma mudança significativa ocorreu da linha de base até a semana 52 (alteração vs placebo: -0,3 pontos de 236 possíveis no grupo de 250 mg, 0,5 pontos no grupo de 1.250 mg e 0,1 ponto no grupo de 3.500 mg) ou em semana 72 (diferença no início do cinpanemab na linha de base vs semana 52: -0,9, 0,6 e -0,8 pontos, respectivamente).
No estudo PASADENA, o prasinezumabe também não alterou significativamente o mesmo desfecho da linha de base até a semana 52 (alteração vs placebo: -2,0 pontos no grupo de 1.500 mg e -0,6 pontos no grupo de 4.500 mg), relatou Gennaro Pagano, MD , PhD, do Roche Innovation Center Basel, na Suíça, e colegas. Uma coorte de início tardio produziu resultados igualmente negativos.
O efeito não foi significativo em geral para as medidas de imagem, concluíram ambos os grupos nos estudos publicados juntos no New England Journal of Medicine.
Os resultados foram "mais do que decepcionantes e certamente não têm implicações para a prática atual", observou Alan Whone, PhD, da Universidade de Bristol e Southmead Hospital em Bristol, Inglaterra, em um editorial de acompanhamento.
“Os dados negativos aparentemente não impediram o patrocinador do PASADENA de iniciar um estudo de fase 2b, embora pareça provável que a evidência agregada marque o fim do caminho para anticorpos monoclonais no tratamento da doença de Parkinson inicial”, escreveu ele.
A Biogen anunciou que estava descontinuando o desenvolvimento de cinpanemab.
Ambos
os agentes investigacionais têm como alvo a forma agregada da
proteína alfa-sinucleína, considerada a principal culpada na
patogênese da doença de Parkinson. Ambos são um anticorpo
monoclonal humanizado que se liga seletivamente à alfa-sinucleína
agregada, que o prasinezumab agarra no C-terminal da proteína e o
cinpanemab no N-terminal.
O prasinezumab teve um benefício
significativo em um desfecho secundário - progressão mais lenta com
a dose mais baixa na parte III do MDS-UPDRS, refletindo o exame motor
conduzido pelo médico, observou o grupo de Pagano.
Whone
advertiu que os desfechos secundários não foram corrigidos para
comparações múltiplas, "e, portanto, nenhuma conclusão pode
ser tirada deles".
"Ainda assim, isso não deve
descartar as possibilidades de que o sucesso ainda possa ser
alcançado com os mesmos ou semelhantes agentes na doença de
Parkinson prodrômica ou em formas genéticas do distúrbio ou que
mecanismos alternativos para afetar a alfa-sinucleína agregada
possam ser benéficos", escreveu ele.
Uma questão maior
que Whone levantou é se a "lamentável falta de realização"
dos agentes modificadores da doença para a doença de Parkinson se
deve a pesquisas pré-clínicas enganosas, projetos de ensaios
clínicos atuais que entregam erros do tipo II ou ambos.
"Para
PASADENA e o estudo SPARK, parece que a primeira explicação é mais
provável, mas a última continua sendo possível; se for verdade,
isso pode implicar que as medidas de resultado devem ser mais
sofisticadas e passar para a era digital", escreveu ele.
O
SPARK incluiu 357 pacientes com doença de Parkinson inicial
randomizados para placebo IV ou cinpanemab na dose de 250 mg, 1.250
mg ou 3.500 mg a cada 4 semanas por 52 semanas, seguido por um
período de extensão sem dose de tratamento ativo por até 112
semanas .
O estudo PASADENA envolveu 316 pacientes com doença
de Parkinson em estágio inicial randomizados para receber placebo IV
ou prasinezumabe na dose de 1.500 mg ou 4.500 mg a cada 4 semanas por
52 semanas. A parte 2 do estudo envolveu uma coorte de início tardio
recebendo placebo nas primeiras 52 semanas que mudou para
prasinezumabe na dose de 1.500 ou 4.500 mg das semanas 56 a
104.
Ambos os ensaios envolveram pacientes na América do
Norte e na Europa, com a adição de Israel no SPARK.
Apesar
dos resultados negativos, escreveu Whone, "se alguém quiser
tentar novamente do zero - e ainda é possível que haja um atraso
temporal entre a eliminação da alfa-sinucleína agregada e a
preservação neuronal e que uma duração consideravelmente maior do
teste pode ser mais bem sucedido - ambos os agentes pareciam ser
relativamente seguros e não suscitaram preocupações de
imunogenicidade". Original em inglês, tradução Google,
revisão Hugo. Fonte: Medpagetoday.
sexta-feira, 22 de julho de 2022
Oportunidades e desafios da alfa-sinucleína como um potencial biomarcador para a doença de Parkinson e outras sinucleinopatias
22 July 2022 - Resumo - A doença de Parkinson (DP), a segunda doença neurodegenerativa progressiva mais comum, desenvolve-se e progride por 10 a 15 anos antes que os sintomas diagnósticos clínicos da doença se manifestem. Além disso, vários aspectos da patologia da DP se sobrepõem a outras doenças neurodegenerativas (NDDs) ligadas à agregação de alfa-sinucleína (aSyn), também chamadas de sinucleinopatias. Portanto, há uma necessidade urgente de descobrir e validar marcadores diagnósticos e prognósticos precoces que reflitam a fisiopatologia da doença, progressão, gravidade e diferenças potenciais nos mecanismos da doença entre DP e outros NDDs. A estreita associação entre aSyn e o desenvolvimento de patologia em sinucleinopatias, juntamente com a identificação de espécies de aSyn em fluidos biológicos, levou a um crescente interesse em espécies de aSyn como potenciais biomarcadores para diagnóstico precoce de DP e diferenciá-lo de outras sinucleinopatias. Nesta revisão, nós (1) fornecemos uma visão geral do progresso em direção ao mapeamento da distribuição de espécies aSyn no cérebro, tecidos periféricos e fluidos biológicos; (2) apresentar análise comparativa e crítica de estudos anteriores que mediram aSyn total, bem como outras espécies, como formas modificadas e agregadas de aSyn em diferentes fluidos biológicos; e (3) destacar lacunas e desafios conceituais e técnicos que podem dificultar o desenvolvimento e validação de biomarcadores aSyn confiáveis; e (4) delinear uma série de recomendações para enfrentar esses desafios. Finalmente, propomos uma abordagem combinada de biomarcadores baseada na integração de características bioquímicas, de agregação e estrutura de aSyn, além de outros biomarcadores de neurodegeneração. Acreditamos que capturar a diversidade de espécies de aSyn é essencial para desenvolver ensaios e diagnósticos robustos para detecção precoce, estratificação de pacientes, monitoramento da progressão da doença e diferenciação entre sinucleinopatias. Isso pode transformar o design e a implementação de ensaios clínicos, acelerar o desenvolvimento de novas terapias e melhorar as decisões clínicas e as estratégias de tratamento.
Introdução
A doença de
Parkinson (DP) é uma das doenças neurodegenerativas (NDDs) mais
progressivas, com uma taxa de prevalência mundial de ~1-4% em
pessoas com mais de 60 anos1. Espera-se que a incidência da DP
aumente como resultado da maior expectativa de vida2. A DP é
caracterizada pela perda progressiva de neurônios dopaminérgicos e
pela deposição de alfa-sinucleína agregada (aSyn) em inclusões
intracelulares que se acumulam na forma de corpos de Lewy (LBs) nos
corpos celulares e neuritos de Lewy (LNs) nos axônios e dendritos3.
Até o momento, o diagnóstico clínico da DP tem sido baseado em
características motoras, juntamente com sintomas não motores, como
características psiquiátricas e autonômicas e distúrbios do
sono4,5,6,7. A detecção da patologia aSyn no cérebro post-mortem
continua a ser o principal meio de chegar a um diagnóstico
conclusivo, muitas vezes revelando que casos significativos de DP
foram diagnosticados erroneamente8. Como o diagnóstico de DP se
baseia em sintomas clínicos que se manifestam apenas após uma perda
substancial e irreversível de neurônios dopaminérgicos na
substância negra (SN), há uma necessidade urgente de identificar
biomarcadores específicos de DP que permitam o diagnóstico no
início e/ou estágios iniciais da doença9. Além disso, dada a
sobreposição clínica e neuropatológica entre DP e outras
sinucleinopatias (por exemplo, demência com corpos de Lewy (DLB) e
atrofia de múltiplos sistemas (MSA)), há também a necessidade de
biomarcadores que permitam a diferenciação entre sinucleinopatias.
A descoberta de marcadores diagnósticos e prognósticos precoces que
refletem a fisiopatologia, progressão e gravidade da doença e
refletem diferenças potenciais nos mecanismos da doença são de
suma importância e são uma grande promessa para melhorar o desenho
de ensaios clínicos e o desenvolvimento de novas ferramentas e
terapias de diagnóstico específicas da doença para DP e outras
sinucleinopatias. Original em inglês, tradução Google, revisão
Hugo. Fonte: Nature.
Assinar:
Postagens (Atom)